Classical MD Modules¶
Introduction¶

This is a collection of the modules that have been created by E-CAM community within the area of Classical MD. This documentation is created using ReStructured Text and the git repository for the documentation source files can be found at https://gitlab.e-cam2020.eu/e-cam/E-CAM-Library which are open to contributions from E-CAM members.
In the context of E-CAM, the definition of a software module is any piece of software that could be of use to the E-CAM community and that encapsulates some additional functionality, enhanced performance or improved usability for people performing computational simulations in the domain areas of interest to us.
This definition is deliberately broader than the traditional concept of a module as defined in the semantics of most high-level programming languages and is intended to capture inter alia workflow scripts, analysis tools and test suites as well as traditional subroutines and functions. Because such E-CAM modules will form a heterogeneous collection we prefer to refer to this as an E-CAM software repository rather than a library (since the word library carries a particular meaning in the programming world). The modules do however share with the traditional computer science definition the concept of hiding the internal workings of a module behind simple and well-defined interfaces. It is probable that in many cases the modules will result from the abstraction and refactoring of useful ideas from existing codes rather than being written entirely de novo.
Perhaps more important than exactly what a module is, is how it is written and used. A final E-CAM module adheres to current best-practice programming style conventions, is well documented and comes with either regression or unit tests (and any necessary associated data). E-CAM modules should be written in such a way that they can potentially take advantage of anticipated hardware developments in the near future (and this is one of the training objectives of E-CAM).
Rare events and path sampling¶
In many simulations, we come across the challenge of bridging timescales.
The desire for high resolution in space (and therefore time) is inherently
in conflict with the desire to study long-time dynamics. To study molecular
dynamics with atomistic detail, we must use timesteps on the order of a
femtosecond. However, many problems in biological chemistry, materials
science, and other fields involve events that only spontaneously occur after
a millisecond or longer (for example, biomolecular conformational changes,
or nucleation processes). That means that we would need around
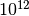 time steps to see a single millisecond-scale event. This is
the problem of “rare events” in theoretical and computational chemistry.
time steps to see a single millisecond-scale event. This is
the problem of “rare events” in theoretical and computational chemistry.
While modern supercomputers are beginning to make it possible to obtain trajectories long enough to observe some of these processes (such as millisecond dynamics of a protein), even then, we may only find one example of a given transition. To fully characterize a transition (with proper statistics), we need many examples. This is where path sampling comes in. Path sampling approaches obtain many trajectories using a Markov chain Monte Carlo approach: An existing trajectory is perturbed (usually using a variant of the “shooting” move), and the resulting trial trajectory is accepted or rejected according to conditions that preserve the distribution of the path ensemble. As such, path sampling is Monte Carlo in the space of paths (trajectories). Conceptually, this enhances the sampling of transitions by focusing on the transition region instead of the stable states. In direct MD, trajectories spend much more time in stable states than in the transition region (exponential population differences for linear free energy differences); path sampling skips over that time in the stable states.
The main path sampling approaches used in the modules below are transition path sampling (TPS) and transition interface sampling (TIS). In practice, TPS is mainly used to characterize the mechanism of a transition, while TIS (which is more expensive than TPS) is used to calculate rates and free energy landscapes. Overviews of these methods, as well as other rare events methods, can be found in the following review articles:
- 2010 review by Bolhuis and Dellago in Reviews in Computational Chemistry
- 2008 review by Dellago and Bolhuis in Advances in Polymer Science
In addition, several other resources are available on the web to teach path sampling, including:
Since the problem of bridging timescales, which path sampling addresses, is a generic one, path sampling can be used in many fields. Indeed, there’s nothing in the methodology that even restricts it to molecular simulation. However, it is best known in the field of classical MD simulations, where path sampling methods have shown many successes, including:
- Mechanisms of complex chemical reactions, such as autoionization of water
- Mechanism of hydrophobic assembly
- Evidence that the glass transition is a first-order phase transition
- Mechanism of crystal nucleation
- Mechanism of cavitation
- Identifying new mechanisms in catalytic systems
- Characterization of the conformational dynamics networks in proteins
As computational resources become more powerful, path sampling has the promise to provide insight into rare events in larger systems, and into events with even longer timescales. For example:
- Drug/protein binding and unbinding (timescales of minutes), which is essential for predicting the efficacy of drugs
- Association processes of proteins (large systems), which is at the core of communication in biochemical pathways
- Self assembly processes for complex systems (many intermediates), which can be important for the design of new materials
Further, applying the known successes of path sampling methods to larger systems can also be quite valuable. Path sampling can shed light on the networks of conformational dynamics for large proteins and protein complexes, and on the mechanisms and rates of complex reactions and phase transitions. The range of possibilities is so broad that it is impossible to enumerate – both academics and industry will benefit greatly from having software for these methods.
The modules listed here deal with software to perform path sampling methods, as well as other approaches to rare events.
OpenPathSampling¶
Several modules were developed based on OpenPathSampling (OPS). These include modules that have been incorporated into the core of OPS, as well as some that remain separate projects. The modules that were incorporated into the core are:
- Path Density for OpenPathSampling
- Direct MD (on-the-fly) flux/rate in OpenPathSampling
- Improved input for OPS networks
- New WHAM code
- Flux/Rate Analysis in OpenPathSampling
- OpenPathSampling Snapshot Features
- Two-Way Shooting in OpenPathSampling
- Committor Analysis in OpenPathSampling
- OPS Channel Analysis
- OPS New TIS Analysis
- Resampling Statistics
- Gromacs engine in OpenPathSampling
- OPS Visit All States Ensemble
- Interface-Constrained Shooting in OpenPathSampling
- Progress meters in OPS analysis
- Faster Path Density Analysis in OPS
- SimStore: OPS New Storage Subsystem (part 1)
- SimStore: Storable Functions
- OPS New Storage Subsystem
- SimStore: Support for OpenMM Snapshots
The modules that are based on OPS, but remain separate, are:
Nine of these modules were part of E-CAM Deliverable 1.2. Those modules provided improvements and new features in software for trajectory sampling and for studying the thermodynamics and kinetics of rare events.
Machine Learning Potentials¶
Many systems in computational physics and chemistry can be successfully studied with empirical force fields at the atomistic level. In the context of these “molecular mechanics” models, atoms are treated as particles without internal structure and their interactions are defined via rather simple expressions deduced from physical/chemical intuition. Usually a small number of free parameters is enough to tune the potential to reproduce experimental properties with good agreement. However, there are systems for which a satisfying description within this framework is not possible. Take as an example the formation and breaking of covalent bonds. This is the territory of ab initio methods which use quantum mechanics to accurately model the behavior of the system. Unfortunately the additional level of detail comes at a cost. Even in small systems ab initio methods are usually many orders of magnitude slower than empirical force fields. Moreover, the computational cost increases unfavorably with the number of atoms which makes it impractical to perform large simulations.
With rising influence of machine learning algorithms in science and technology a new category of interatomic potentials has emerged. Machine learning potentials (MLPs) aim at bridging the gap between ab initio methods and empirical force fields. In contrast to the latter, MLPs are not bound by a predetermined fixed functional form of the interaction but rather build on the flexibility of an underlying machine learning model, such as artificial neural networks. These are known for their capability to reproduce any complicated function, which in this case is the desired potential energy surface, but rely on a separate training stage before they are ready for use. During this phase the MLP “learns” from a large data set how energies and forces depend on atomic positions. The reference energy landscape is typically computed from expensive ab initio methods. Once the training is completed the MLP can accurately predict energies and forces for new (unseen during training) atomic configurations at a fraction of the cost of the reference method. Hence, with MLPs times scales become accessible in molecular dynamics simulations close to those of empirical potentials while maintaining the ab initio level of accuracy.
Today MLPs exist in various forms and combine different atomic environment descriptors as inputs for all kinds of machine learning models.
A very successful variant is the high-dimensional neural network potential (HDNNP) which combines make use of artificial neural networks to predict atomic energy contributions:
n2p2¶
The software n2p2 (NeuralNetworkPotentialPackage) implements the HDNNP method in a C++ library and applications for training and prediction. Its most important feature in the HPC context is the interface to the popular molecular dynamics package LAMMPS which allows to use HDNNPs in massively parallelized simulation runs. Further information can be found in these two publications:
The following modules extend the functionality of n2p2, some are already merged into the main repository, others will also work independently and will be integrated in the future:
Pilot Projects¶
One of primary activity of E-CAM is to engage with pilot projects with industrial partners. These projects are conceived together with the partner and typically are to facilitate or improve the scope of computational simulation within the partner. The related code development for the pilot projects are open source (where the licence of the underlying software allows this) and are described in the modules associated with the pilot projects.
More information on Classical MD pilot projects can be found on the main E-CAM website:
The following modules were developed specifically for the Classical MD pilot projects.
- Contact Map
- Contact Map Parallelization
- Contact Concurrences
- Particle Insertion Core
- Particle Insertion Hydration
- LAMMPS-pyinterfaceExt
- E-CAM minDist2segments_KKT module
- E-CAM minDist2segments_KKT_for_SRP module
- E-CAM velocities_resolve_EVC module
- E-CAM velocities_resolve_EVC_for_LAMMPS module
- E-CAM
Verlet_list_for_ODEmodule - E-CAM openmm_copolymer module
- E-CAM openmm_plectoneme module
- E-CAM polymer_data_analysis module
- E-CAM 2spaces_on_gpu module
- n2p2 - CG descriptor analysis
- n2p2 - Improved link to HPC MD software
- n2p2 - Polynomial Symmetry Functions
- n2p2 - Symmetry Function Memory Footprint Reduction
Extended Software Development Workshops (ESDWs)¶
The first ESDW for the Classical MD workpackage was held in Traunkirchen, Austria, in November 2016, with a follow-up to be held in Vienna in April 2017. The following modules have been produced:
The second ESDW for the Classical MD workpackage was held in Leiden, Holland, in August 2017. The following modules have been produced:
The third ESDW for the Classical MD work package was held in Turin, Italy in July 2018. The following have been produced as a result:
ESDW Lyon 2019¶
ESDW Clifden 2019¶
The ESDW on “Inverse Molecular Design & Inference: building a Molecular Foundry” in Clifden, Ireland in November 2019 was the starting point for the modules below.
European Environment for Scientific Software Installations¶
A number of modules related to the E-CAM support of the European Environment for Scientific Software Installations EESSI which is is a collaboration between a number of academic and industrial partners in the HPC community. Through the EESSI project, they want to set up a shared stack of scientific software installations to avoid not only duplicate work across HPC sites but also the execution of sub-optimal applications on HPC resources.
For end users, EESSI wants to provide a uniform user experience with respect to available scientific software, regardless of which system they use. The software stack is intended to work on laptops, personal workstations, HPC clusters and in the cloud, which means the project will need to support different CPUs, networks, GPUs, and so on. The intention is to make this work for any Linux distribution, and a wide variety of CPU architectures (Intel, AMD, ARM, POWER, RISC-V).
The pilot instance of the EESSI software stack includes GROMACS, and benchmarking is being done by E-CAM using that application, which is why we include these modules in this section.