PaPIM¶
Purpose of Module¶
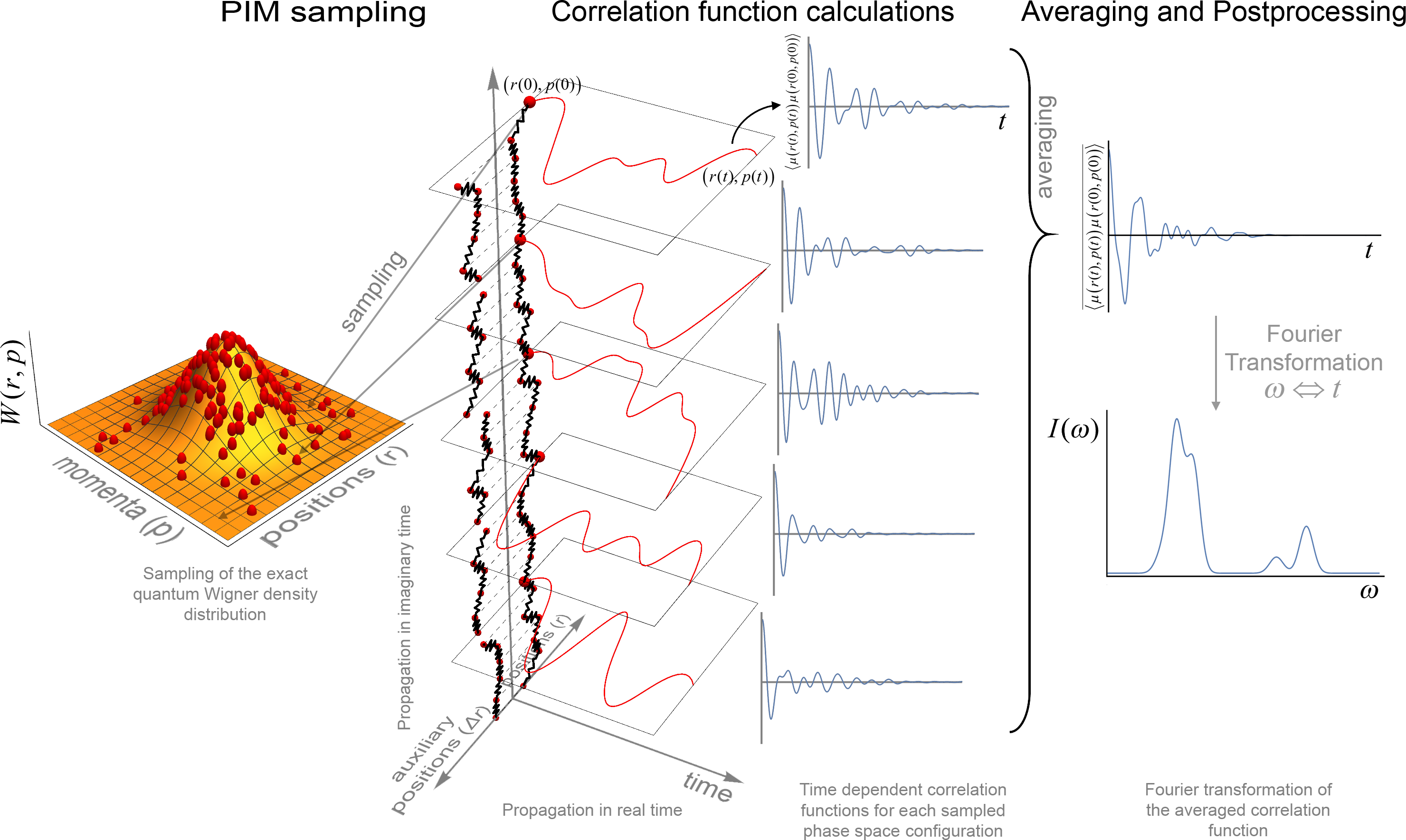
PaPIM is a code for computing time-dependent correlation functions and sampling of the phase space.
It samples the phase space either classically or quantum mechanically.
For the classical sampling of the phase space a Monte Carlo algorithm samples the Boltzmann distribution function,
while for the quantum sampling a Phase Integration Method (PIM) [PMon1] [PMon2] is utilized for an exact sampling of the quantum
Wigner density distribution.
From the sampled phase space points trajectories are propagated in time using classical molecular dynamics
in order to obtain the appropriate time-dependent correlation functions.
The code is designed so the user can easily couple it with its own external potential energy code/library
and/or correlation functions subroutines.
Examples for the 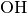 ,
, 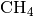 and
and 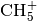 systems are provided,
where for the system an external subroutine for calculation of
system potential energy and electric dipole moment, based on fitted values, [PJin] is also provided.
The first two systems use Morse and harmonic potential, respectively.
PaPIM comes with a direct interface to the CP2K program package program package for
calculation of system’s electronic structure properties.
The CP2K package is coupled to PaPIM as a library, thus avoiding the exchange of information process via writing and
reading to an external file.
For more information see the corresponding PaPIM-CP2K_Interface module.
.. Example external subroutines are provided for the and systems, respectively,
.. with potential energies described by the harmonic potential,
.. and the electric dipole moments by point charge approximation.
.. An external subroutine for calculation of
.. system potential energy and electric dipole moment, based on fitted values, [PJin] is also given.
The electric dipole moment operator is currently implemented into the code for calculation of the
electric dipole moment autocorrelation function from which system IR spectra can be directly obtained.
systems are provided,
where for the system an external subroutine for calculation of
system potential energy and electric dipole moment, based on fitted values, [PJin] is also provided.
The first two systems use Morse and harmonic potential, respectively.
PaPIM comes with a direct interface to the CP2K program package program package for
calculation of system’s electronic structure properties.
The CP2K package is coupled to PaPIM as a library, thus avoiding the exchange of information process via writing and
reading to an external file.
For more information see the corresponding PaPIM-CP2K_Interface module.
.. Example external subroutines are provided for the and systems, respectively,
.. with potential energies described by the harmonic potential,
.. and the electric dipole moments by point charge approximation.
.. An external subroutine for calculation of
.. system potential energy and electric dipole moment, based on fitted values, [PJin] is also given.
The electric dipole moment operator is currently implemented into the code for calculation of the
electric dipole moment autocorrelation function from which system IR spectra can be directly obtained.
Phase Integration Method (PIM)¶
The Phase Integration Method (PIM) is a novel approximate quantum dynamical technique developed for computing systems time dependent observables. [PMon1] [PMon2] [PBeu] PIM employs an algorithm in which the exact sampling of the quantum thermal Wigner density is combined with a linearized approximation of the quantum time propagators represented in the path integral formalism that reduces the evolution to classical dynamics. The quantities of interest can then be computed by combining classical molecular dynamics algorithms with a generalized Monte Carlo sampling scheme for sampling of the quantum initial conditions.
Applications of the Module¶
The PaPIM code has been extensively used for the calculation of the system infrared absorption
spectrum in the gas phase.
These calculations also provided the benchmark of the PIM method as well as for the code performance analysis.
The results obtained on the system are currently under preparation for publication.
One master thesis was completed by applying the code.
Investigations of the processes shaping the infrared spectrum of small water cluster systems and a protoneted
water dimer system are currently being investigated using the PaPIM code.
Compiling¶
Fortran compiler with a MPI wrapper together with lapack libraries have to be available to successfully compile the code.
The user is advised to examine the Makefile in the ./source sub-directory prior to code compilation in order to
select an appropriate compiler and to check or adapt the compiler options to his local environment, or to generally
modify the compiler options to his requirements.
Upon adapting the Makefile, the code compilation is executed by command make in the ./source
sub-directory.
The executable PaPIM.exe is created at the same location upon successful compilation.
For module’s testing purposes the user is advise to have numdiff package installed before running the tests.
More details on the numdiff program package and its installation is available
here.
For compiling the PaPIM with an interface to the CP2K package see here.
The PaPIM documentation is generated by executing the make command in the ./doc sub-directory.
Testing¶
Testing check and validates the successful compilation of the PaPIM code.
Tests and all corresponding reference files are located in sub-directory ./tests.
If using the automated testing script all tests have to be performed in this sub-directory.
The tests are performed on three systems, the , and .
They are located in their corresponding sub-directories, oh, ch4 and ch5, where each sub-directory
contains corresponding classical and quantum input files located in CLASSICAL and QUANTUM sub-directories,
respectively.
Tests are performed automatically in the ./tests sub-directory by executing the command:
./test.sh -n [number of cores]
Flag -n [number of cores] controls the number of processor cores used in the tests.
By default the tests are performed on two processor cores, which can be changed by setting a different number of
required processor cores.
Because the number of trajectories for sampling in certain tests are limited to 20, the number
of processor cores should not exceed 20.
For testing of the PaPIM code linked with the CP2K package see here.
For comparison of generated output values with reference data the test script uses numdiff command
in order to compensate for small numerical differences.
By default the script looks first for the numdiff command on the system, and in case it fails to
locate it, the standard diff command will be used instead.
However, the user is warned that due to small numerical differences between generated output and corresponding
reference values the automated tests are most likely to fail.
A local numdiff package copy can be included in the test by specifying its absolute path.
For this and other options of the test script list them with the command ./test.sh -h.
Performance and Benchmarking¶
PaPIM is designed as a highly scalable code. Its performance was extensively tested.
Source Code¶
The PaPIM module source code can be obtained from: https://gitlab.e-cam2020.eu:10443/Quantum-Dynamics/PIM/tree/master/source.
Source Code Documentation¶
The source code documentation is located in the ./doc sub-directory.
The documentation files (html and latex format) are generated by executing the make command in the ./doc
sub-directory.
References¶
| [PMon1] | (1, 2) M. Monteferrante, S. Bonella, G. Ciccotti Mol. Phys. 109 (2011) 3015 DOI: 10.1080/00268976.2011.619506 |
| [PMon2] | (1, 2) M. Monteferrante, S. Bonella, G. Ciccotti J. Chem. Phys. 138 (2013) 054118 DOI: 10.1063/1.4789760 |
| [PBeu] | J. Beutier, M. Monteferrante, S. Bonella, R. Vuilleumier, G. Ciccotti Mol. Sim. 40 (2014) 196 DOI: 10.1080/08927022.2013.843776 |
| [PJin] | (1, 2) Z. Jin, B. Braams, J. Bowman J. Phys. Chem. A 110 (2006) 1569 DOI: 10.1021/jp053848o |