PIM_wd¶
Purpose of Module¶
Module PIM_wd implements the Phase Integration Method (PIM) [Mon1] [Mon2] for the exact sampling of the quantum Wigner distribution in phase space representation. The PIM samples the thermal Wigner density using a generalized Monte Carlo scheme for sampling phase space points. The scheme combines the Penalty [Pen] and Kennedy [Ken] algorithms to sample noisy probability densities. This is necessary because the estimator of the quantum thermal density is not known analytically but must be computed via a statistical average affected by uncertainty. The sampled points are the basis for the calculation of time-dependent correlation function with the PIM algorithm via the module PaPIM. The user is required to provide the potential energy of the system by incorporating an external potential energy subroutine into the PotMod potential energy library.
Applications of the Module¶
This module forms the basis for computing the time-dependent cross- and auto-correlation functions with the PIM algorithm.
It has been used in the calculation of 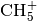 infrared spectrum and in the gas phase as well as for the
computation of infrared spectrum of small water molecule clusters and protonated water dimer system.
infrared spectrum and in the gas phase as well as for the
computation of infrared spectrum of small water molecule clusters and protonated water dimer system.
Compiling¶
Fortran compiler with a MPI wrapper together with lapack libraries have to be available to successfully compile the code.
The user is advised to examine the Makefile in the ./source` sub-directory prior to code compilation in order to
select an appropriate compiler and to check or adapt the compiler options to his local environment, or to generally
modify the compiler options to his requirements.
cd source
make
Upon adapting the Makefile, the code compilation is executed by command make in the ./source sub-directory.
An executable PaPIM.exe is created upon successful compilation.
Testing¶
For PIM_wd test purposes the numdiff package is used for automatic comparison purposes and should be made
available before running the tests, otherwise the diff command will be used automatically instead but the user
is warned that the test might fail due to numerical differences.
The user is advised to download and install numdiff from here.
Tests and corresponding reference values are located in sub-directories ./tests/. The tests are performed over
three systems, the 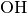 ,
, 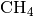 and . They are located in their corresponding
and . They are located in their corresponding
oh, ch4 and ch5,
where each sub-directory contains corresponding classical and quantum input files located in CLASSICAL and QUANTUM
sub-directories, respectively.
Before running the tests the code has to be properly compiled by running the make command in the
./source sub-directory.
The tests are performed automatically by executing the command ./test.sh in the ./tests sub-directory
for all three systems:
cd tests
./test.sh [number of cores]
Tests are by default performed using two processor cores, which can be changed by setting the value of required
cores as an integer number after the command ./test.sh (example ./test.sh 20, for the use of 20 processor
cores in the test).
The number of processor cores should not exceed 20.
Due to small numerical discrepancies between generated outputs and reference values which can cause the tests to fail,
the user is advised to manually examine the numerical differences between generated output and the corresponding
reference values in case the tests fail.
Source Code¶
The PIM_qcf module source code is located at: https://gitlab.e-cam2020.eu:10443/Quantum-Dynamics/PIM/tree/PIM_wd.
Source Code Documentation¶
The source code documentation can be generated automatically in ./doc sub-directory,
html and latex format, by executing the following command in the ./doc directory:
doxygen PIMwd_doxygen_settings
References¶
| [Pen] | D. M. Ceperley, M. Dewing J. Chem. Phys. 110 (1999) 9812 DOI: http://dx.doi.org/10.1063/1.478034 |
| [Ken] | A. D. Kennedy, J. Kuti Phys. Rev. Lett. 54 (1985) 2473 DOI: https://doi.org/10.1103/PhysRevLett.54.2473 |