Analysis of local tetrahedral ordering for DL_MESO_DPD¶
Purpose of Module¶
This module, tetrahedral.f90, is a postprocessing utility for DL_MESO_DPD,
the Dissipative Particle Dynamics (DPD) code from the DL_MESO package.
It processes trajectory (HISTORY) files and analyzes the local tetrahedral
ordering, a feature that is relevant, for example, in water-like systems.
The local ordering in liquid water can be assessed considering the coordinates of oxygen atoms [Duboue2015]. In particular, for each oxygen, its four nearest neighbouring oxygens are considered, whereas the hydrogens are disregarded. At the mesoscale level, the user will select one (appropriate) bead species and analyze its local ordering.
Given a particle 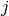 , we first find its four nearest neighbours
(n.n.). Then, an orientational tetrahedral order parameter is built using
, we first find its four nearest neighbours
(n.n.). Then, an orientational tetrahedral order parameter is built using
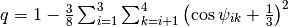 ,
where
,
where 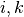 are n.n. of and
are n.n. of and 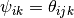 is the angle [1]
between the particles
is the angle [1]
between the particles 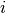 , and
, and 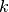 .
Of course, the quantity is then averaged over the central particle and over time.
.
Of course, the quantity is then averaged over the central particle and over time.
A translational tetrahedral order parameter, 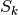 , is defined as
, is defined as
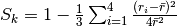 ,
where is a n.n. of and
,
where is a n.n. of and 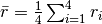 .
.
Concerning the limiting values of these parameters:
in a regular tetrahedron (if the four vertices are referred to the
center of the solid) one has 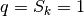 .
In an ideal gas, where the angle
.
In an ideal gas, where the angle 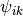 is randomly distributed,
is randomly distributed,
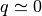 . On the other hand,
. On the other hand, 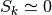 if the density
fluctuations are large enough.
if the density
fluctuations are large enough.
As a result of the analysis, a file TETRADAT is produced, whose columns are
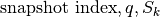 , the instantaneous values of the order
parameters defined above. At the end of the file, the averages and
standard errors (computed assuming the snapshots are uncorrelated) of both order parameters are given.
, the instantaneous values of the order
parameters defined above. At the end of the file, the averages and
standard errors (computed assuming the snapshots are uncorrelated) of both order parameters are given.
Background Information¶
The base code for this module is DL_MESO_DPD, the Dissipative Particle
Dynamics code from the mesoscopic simulation package DL_MESO,
developed by M. Seaton at Daresbury Laboratory.
This open source code is available from STFC under both academic (free) and
commercial (paid) licenses. The module is to be used with DL_MESO
in its latest released version, version 2.7 (released December 2018).
A variant of this module to be used with a previous version of DL_MESO,
version 2.6 (dating November 2015), can be found in the old-v2.6
directory.
Testing¶
The utility tetrahedral.f90 is compiled with the available
Fortran 2003 compiler [2], e.g.:
gfortran -o tetrahedral.exe tetrahedral.f90
and the executable must be in the same directory of the HISTORY file.
The user is asked to provide the number of the species for which ordering has to be
analyzed. To input the user-defined parameter, one can enter it interactively
at runtime or write it into a text file (say, input.txt) and run the program
in this way:
tetrahedral.exe < input.txt
Below we propose a test where a fluid is prepared in a ordered configuration (diamond cubic lattice) and rapidly goes into an orientationally disordered one.
Test
The sources used for this test are available to download.
Run the DL_MESO_DPD simulation on a single node (serial run) using the CONTROL file,
One species - starting as diamond cubic lattice
#volume 64.0
temperature 1.0
cutoff 1.0
timestep 0.01
steps 1000
traj 0 10 0
#traj 500 100 0
stats every 100
stack size 100
print every 100
job time 7200.0
close time 100.0
ensemble nvt mdvv
conf zero
nfold 2,2,2
finish
the FIELD file
One species - starting as diamond cubic lattice
SPECIES 1
A 1.0 0.0 8 0
INTERACTIONS 1
A A dpd 25.0 1.0 4.0
CLOSE
and the CONFIG file
One species - starting as diamond cubic lattice
0 1
4.0000000000 0.0000000000 0.0000000000
0.0000000000 4.0000000000 0.0000000000
0.0000000000 0.0000000000 4.0000000000
A 1
0.0 0.0 0.0
A 2
0.0 2.0 2.0
A 3
2.0 0.0 2.0
A 4
2.0 2.0 0.0
A 5
3.0 3.0 3.0
A 6
3.0 1.0 1.0
A 7
1.0 3.0 1.0
A 8
1.0 1.0 3.0
This configuration corresponds to a diamond cubic lattice [3], while the nfold
directive in the CONTROL file replicates the configuration twice in all three dimensions.
Analyzing the resulting trajectory (HISTORY) file with tetrahedral.exe
(compiled as indicated above) and inputing 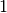 for the runtime argument,
the following output is printed to the standard output:
for the runtime argument,
the following output is printed to the standard output:
Species 1 : A
Which species number has to be analyzed?
1
total number of beads: 64
number of beads by species: 64
number of analyzed beads: 64
<q> = 0.132069E+00
error = 0.159218E-01
<s_k> = 0.986906E+00
error = 0.243544E-03
The output file TETRADAT
# Local ordering for beads of species: A
# dimx, dimy, dimz= 0.0000000000000000 0.0000000000000000 0.0000000000000000
# snapshot number, q, sk
1 1.000000E+00 1.000000E+00
2 9.690709E-01 9.982591E-01
3 8.476516E-01 9.935607E-01
4 5.326193E-01 9.892793E-01
5 3.297230E-01 9.868977E-01
6 2.057773E-01 9.847919E-01
7 9.033514E-02 9.829358E-01
8 2.481253E-02 9.827499E-01
9 4.596717E-02 9.831773E-01
10 4.275300E-02 9.840823E-01
contains the values of 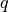 and for each snapshot and their
averages are also produced.
and for each snapshot and their
averages are also produced.
One can see that in the initial snapshot, both order parameters detect an
ordered state (i.e., 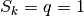 ).
With the evolution in time, since the system is a dilute fluid without bonds
between particles, the orientational ordering is rapidly lost (i.e.,
).
With the evolution in time, since the system is a dilute fluid without bonds
between particles, the orientational ordering is rapidly lost (i.e., 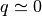 ).
On the other hand, the translational order parameter stays close to one since the density of the
system is roughly uniform.
).
On the other hand, the translational order parameter stays close to one since the density of the
system is roughly uniform.
Source Code¶
You can directly download the source file tetrahedral.f90
and we also include its contents below (as well as in the test tarball).
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184 185 186 187 188 189 190 191 192 193 194 195 196 197 198 199 200 201 202 203 204 205 206 207 208 209 210 211 212 213 214 215 216 217 218 219 220 221 222 223 224 225 226 227 228 229 230 231 232 233 234 235 236 237 238 239 240 241 242 243 244 245 246 247 248 249 250 251 252 253 254 255 256 257 258 259 260 261 262 263 264 265 266 267 268 269 270 271 272 273 274 275 276 277 278 279 280 281 282 283 284 285 286 287 288 289 290 291 292 293 294 295 296 297 298 299 300 301 302 303 304 305 306 307 308 309 310 311 312 313 314 315 316 317 318 319 320 321 322 323 324 325 326 327 328 329 330 331 332 333 334 335 336 337 338 339 340 341 342 343 344 345 346 347 348 349 350 351 352 353 354 355 356 357 358 359 360 361 362 363 364 365 366 367 368 369 370 371 372 373 374 375 376 377 378 379 380 381 382 383 384 385 386 387 388 389 390 391 392 393 394 395 396 397 398 399 400 401 402 403 404 405 406 407 408 409 410 411 412 413 414 415 416 417 418 419 420 421 422 423 424 425 426 427 428 429 430 431 432 433 434 435 436 437 438 439 440 441 442 443 444 445 446 447 448 449 450 451 452 453 454 455 456 457 458 459 460 461 462 463 464 465 466 467 468 469 470 471 472 473 | PROGRAM tetrahedral
!***********************************************************************************
!
! module to analyze tetrahedral ordering in dl_meso HISTORY files
!
! authors - m. a. seaton & s. chiacchiera, january 2018 (tidied up and amended
! january 2021)
!
!**********************************************************************************
IMPLICIT none
INTEGER, PARAMETER :: dp = SELECTED_REAL_KIND (15, 307)
INTEGER, PARAMETER :: si = SELECTED_INT_KIND (8)
INTEGER, PARAMETER :: li = SELECTED_INT_KIND (12)
INTEGER, PARAMETER :: endversion = 1
REAL(KIND=dp), PARAMETER :: pi=3.141592653589793_dp
INTEGER, PARAMETER :: ntraj=10
CHARACTER(80) :: text
CHARACTER(8), ALLOCATABLE :: namspe (:), nammol (:)
INTEGER, ALLOCATABLE :: ltp (:), nspec (:), readint (:)
INTEGER :: nrtout
INTEGER :: chain, ioerror, i, numtraj, j, k, nmoldef, ibond
INTEGER :: nspe, nbeads, nusyst, nsyst, numbond, global, species, molecule
INTEGER :: lfrzn, keytrj, srfx, srfy, srfz
INTEGER :: nav
INTEGER :: bead1, bead2
INTEGER :: endver, Dlen, nstep, framesize, lend
INTEGER(KIND=li) :: filesize
REAL(KIND=dp), ALLOCATABLE :: xxx (:), yyy (:), zzz (:), readdata (:)
REAL(KIND=dp) :: dimx, dimy, dimz, shrdx, shrdy, shrdz
REAL(KIND=dp) :: amass, rcii, chg
REAL(KIND=dp) :: time
LOGICAL :: eof, swapend, bigend
! Variables for tetrahedral ordering
INTEGER :: nnlab(4), npart, sp, count
REAL(KIND=dp) :: qtetra, stetra
REAL(KIND=dp) :: q, sk, q_sum, sk_sum, q_ave, sk_ave
REAL(KIND=dp) :: q2_sum, sk2_sum, q2_ave, sk2_ave
!-----------------------------------------------------------------------------------------
! determine number of bytes for selected double precision kind
! (the default SELECTED_REAL_KIND (15, 307) should return 8 bytes)
lend = STORAGE_SIZE (1.0_dp) / 8
! check endianness of machine
bigend = (IACHAR(TRANSFER(1,"a"))==0)
! Determine if HISTORY file exists, which endianness to use,
! if type of real is correct
INQUIRE (file = 'HISTORY', EXIST = eof)
IF (.NOT. eof) THEN
PRINT *, "ERROR: cannot find HISTORY file"
STOP
END IF
OPEN (ntraj, file = 'HISTORY', access = 'stream', form = 'unformatted', status = 'unknown')
swapend = .false.
READ (ntraj) endver, Dlen
IF (endver/=endversion) THEN
swapend = .true.
CLOSE (ntraj)
IF (bigend) THEN
OPEN (ntraj, file = 'HISTORY', access = 'stream', form = 'unformatted', status = 'unknown', convert = 'little_endian')
ELSE
OPEN (ntraj, file = 'HISTORY', access = 'stream', form = 'unformatted', status = 'unknown', convert = 'big_endian')
END IF
READ (ntraj) endver, Dlen
IF (endver/=endversion) THEN
PRINT *, "ERROR: corrupted HISTORY file or created with incorrect version of DL_MESO"
STOP
END IF
END IF
IF (Dlen/=lend) THEN
PRINT *, "ERROR: incorrect type of real number used in HISTORY file"
PRINT *, " recompile tetrahedral.f90 with reals of ", Dlen, " bytes"
STOP
END IF
! read file size, number of trajectory frames and timestep numbers
READ (ntraj) filesize, numtraj, nstep
! Read the number of beads, molecules and bonds
! Arrays are filled with names of particles and molecules: if checking molecules,
! arrays for species, molecule types etc. also filled
READ (ntraj) text
READ (ntraj) nspe, nmoldef, nusyst, nsyst, numbond, keytrj, srfx, srfy, srfz
ALLOCATE (namspe (nspe), nammol (nmoldef), nspec (nspe)) ! NB: nspec here counts ALL beads of a type, not only unbonded ones
ALLOCATE (xxx (1:nsyst), yyy (1:nsyst), zzz (1:nsyst))
ALLOCATE (ltp (1:nsyst))
framesize = (keytrj+1) * 3
ALLOCATE (readint (1:nsyst), readdata (1:framesize))
DO i = 1, nspe
READ (ntraj) namspe (i), amass, rcii, chg, lfrzn
END DO
DO i = 1, nmoldef
READ (ntraj) nammol (i)
END DO
! Read properties of beads and molecules
nspec (:) = 0 ! populations
ibond = 0 !counter for bonds
DO i = 1, nsyst
READ (ntraj) global, species, molecule, chain
ltp (global) = species
nspec (species) = nspec (species) + 1
END DO
IF (numbond>0) THEN
DO i = 1, numbond
READ (ntraj) bead1, bead2
END DO
END IF
! Find number of beads for which trajectories are needed
DO i = 1, nspe
WRITE(*,*) "Species ",i,": ",namspe (i)
END DO
WRITE(*,*) "Which species number has to be analyzed?"
READ(*,*) sp
IF (sp<0 .OR. sp>nspe) THEN
WRITE(*,*) "error: undefined species!"
STOP
END IF
npart = nspec (sp)
WRITE(*,*) "total number of beads: ", nsyst
WRITE(*,*) "number of beads by species: ", nspec
WRITE(*,*) "number of analyzed beads: ", npart
! Open and write output file
nrtout = ntraj + 1
OPEN (nrtout, file = 'TETRADAT', status ='replace')
WRITE (nrtout,*) "# Local ordering for beads of species: ", namspe (sp)
WRITE (nrtout,*) "# dimx, dimy, dimz=", dimx, dimy, dimz
WRITE (nrtout,*) "# snapshot number, q, sk"
eof = .false.
k = 0
nav = 0
q_sum = 0
sk_sum = 0
q2_sum = 0
sk2_sum = 0
! Read snapshots of trajectories
DO k = 1, numtraj
READ (ntraj, IOSTAT=ioerror) time, nbeads, dimx, dimy, dimz, shrdx, shrdy, shrdz
IF (ioerror/=0) THEN
eof = .true.
IF (k==1) THEN
WRITE (*,*) 'ERROR: cannot find trajectory data in HISTORY files'
STOP
END IF
EXIT
END IF
nav = nav + 1
! The full coordinate arrays are used to avoid re-labelling, but they are filled *only* for particles of species "sp"
xxx (:) = 0.0_dp
yyy (:) = 0.0_dp
zzz (:) = 0.0_dp
count = 0
READ (ntraj) readint (1:nbeads)
DO i = 1, nbeads
global = readint (i)
READ (ntraj) readdata (1:framesize)
IF (ltp (global) == sp) THEN
xxx (global) = readdata (1)
yyy (global) = readdata (2)
zzz (global) = readdata (3)
count = count + 1
END IF
END DO
IF (count /= npart) THEN
WRITE (*,*) " Number of particles of species ",sp," differs from expected!"
STOP
END IF
! ... Analyze the trajectories (snapshot by snapshot) ...
q = 0.0_dp
sk = 0.0_dp
DO i = 1, nsyst
IF (ltp (i) /= sp) CYCLE
CALL closest4 (i, nnlab, npart)
! WRITE (*,*) i, nnlab ! uncomment to see nn labels
CALL compute_tetra_label (i, nnlab, qtetra, stetra)
! print*,"q=",qtetra ! uncomment to print q for each single set of 5 particles
! print*,"s=",stetra ! uncomment to print sk for each single set of 5 particles
q = q + qtetra
sk = sk + stetra
END DO
q = q / REAL(npart, KIND=dp)
sk = sk / REAL(npart, KIND=dp)
WRITE (nrtout,'(1p,I8,2(2x,e14.6))') nav, q, sk
q_sum = q_sum + q
sk_sum = sk_sum + sk
q2_sum = q2_sum + q * q
sk2_sum = sk2_sum + sk * sk
! ...
END DO ! end of loop over trajectories
q_ave = q_sum / REAL(nav, KIND=dp) ! average over snapshots
sk_ave = sk_sum / REAL(nav, KIND=dp)
q2_ave = q2_sum / REAL(nav, KIND=dp) ! average over snapshots
sk2_ave = sk2_sum / REAL(nav, KIND=dp)
WRITE (nrtout,*)
WRITE (nrtout,*)
WRITE (*,'(A9,2x,e14.6)') " <q> = ", q_ave
WRITE (*,'(A9,2x,e14.6)') " error = ", SQRT ((q2_ave - q_ave * q_ave)/REAL(nav, KIND=dp))
WRITE (*,'(A9,2x,e14.6)') " <s_k> = ", sk_ave
WRITE (*,'(A9,2x,e14.6)') " error = ", SQRT ((sk2_ave - sk_ave * sk_ave)/REAL(nav, KIND=dp))
WRITE (nrtout,'(A11,2x,e14.6)') " # <q> = ", q_ave
WRITE (nrtout,'(A11,2x,e14.6)') " # error = ", SQRT ((q2_ave - q_ave * q_ave)/REAL(nav, KIND=dp))
WRITE (nrtout,'(A11,2x,e14.6)') " # <s_k> = ", sk_ave
WRITE (nrtout,'(A11,2x,e14.6)') " # error = ", SQRT ((sk2_ave - sk_ave * sk_ave)/REAL(nav, KIND=dp))
! Close the trajectory file
CLOSE (ntraj)
!close output file
CLOSE (nrtout)
DEALLOCATE (namspe, nammol, nspec)
DEALLOCATE (xxx, yyy, zzz)
DEALLOCATE (ltp)
!-----------------------------------------------------------------------------------------
CONTAINS
SUBROUTINE compute_tetra_label (gb0, nnlab, qtetra, stetra)
!*************************************************************************************
! subroutine to compute q and sk for five particles given their global labels
! (a central one and its four nearest neighbours)
! authors: s. chiacchiera, january 2018
!*************************************************************************************
IMPLICIT none
! NB: I should finally recover the use of subroutine "images"
INTEGER, INTENT(IN):: gb0, nnlab (4)
REAL(KIND=dp), INTENT(OUT) :: qtetra, stetra
REAL(KIND=dp) :: theta, ctheta!, angle_ave, cangle_ave ! can be uncommented for checks
REAL(KIND=dp) :: xab, yab, zab, rab, rrab, xcb, ycb, zcb, rcb, rrcb
REAL(KIND=dp) :: r_ave, r2_ave
INTEGER :: nn1, nn2, i,j,k !change if needed
!-----------------------------------------------------------------------------------------
qtetra = 0.0_dp
! angle_ave = 0._dp
! cangle_ave = 0._dp
j = gb0 ! central particle for angle computations
DO nn1 = 1, 3
i = nnlab (nn1)
DO nn2 =nn1+1, 4
k = nnlab (nn2)
!-----------------------------------------------------------------------------------------
! part to compute the ijk angle (from bond_module.f90)
xab = xxx (i) - xxx (j)
yab = yyy (i) - yyy (j)
zab = zzz (i) - zzz (j)
!!!
! CALL images (xab, yab, zab, dimx, dimy, dimz, srfx, srfy, srfz, shrdx, shrdy, shrdz)
xab = xab - dimx * ANINT (xab/dimx)
yab = yab - dimy * ANINT (yab/dimy)
zab = zab - dimz * ANINT (zab/dimz)
!!!
rab = SQRT(xab * xab + yab * yab + zab * zab)
rrab = MAX (rab, 1e-10_dp)
rrab = 1.0_dp / rrab
xab = xab * rrab
yab = yab * rrab
zab = zab * rrab
xcb = xxx (k) - xxx (j)
ycb = yyy (k) - yyy (j)
zcb = zzz (k) - zzz (j)
!!!
! CALL images (xcb, ycb, zcb, dimx, dimy, dimz, srfx, srfy, srfz, shrdx, shrdy, shrdz)
xcb = xcb - dimx * ANINT (xcb/dimx)
ycb = ycb - dimy * ANINT (ycb/dimy)
zcb = zcb - dimz * ANINT (zcb/dimz)
!!!
rcb = SQRT(xcb * xcb + ycb * ycb + zcb * zcb)
rrcb = MAX (rcb, 1e-10_dp)
rrcb = 1.0_dp / rrcb
xcb = xcb * rrcb
ycb = ycb * rrcb
zcb = zcb * rrcb
ctheta = xab * xcb + yab * ycb + zab * zcb
IF (ABS(ctheta)>1.0_dp) ctheta = SIGN(1.0_dp, ctheta) ! could add a check of how much >1 it is
theta = ACOS (ctheta)
!-----------------------------------------------------------------------------------------
qtetra = qtetra + (ctheta + 1.0_dp/3.0_dp) * (ctheta + 1.0_dp/3.0_dp)
! angle_ave = angle_ave + theta
! cangle_ave = cangle_ave + ctheta
!-----------------------------------------------------------------------------------------
! WRITE(*,'(i2,1x,i2,1x,f13.6,1x,f13.6)') nn1, nn2, ctheta, ACOS(ctheta)/pi*180
END DO
END DO
qtetra = 1.0_dp - 0.375_dp * qtetra
! angle_ave = angle_ave/ 6.
! cangle_ave = cangle_ave/ 6.
! print*,"average angle=", angle_ave
! print*,"average cosine angle=", cangle_ave,"-> angle", ACOS(cangle_ave)," and in degrees ",ACOS(cangle_ave)/pi*180
!-----------------------------------------------------------------------------------------
r_ave = 0.0_dp
r2_ave = 0.0_dp
j = gb0 ! central particle for distance computations
DO nn1 = 1, 4
i = nnlab (nn1)
xab = xxx (i) - xxx (j)
yab = yyy (i) - yyy (j)
zab = zzz (i) - zzz (j)
!!!
! CALL images (xab, yab, zab, dimx, dimy, dimz, srfx, srfy, srfz, shrdx, shrdy, shrdz)
xab = xab - dimx * ANINT (xab/dimx)
yab = yab - dimy * ANINT (yab/dimy)
zab = zab - dimz * ANINT (zab/dimz)
!!!
rab = SQRT(xab * xab + yab * yab + zab * zab)
r_ave = r_ave + rab
r2_ave = r2_ave + rab * rab
END DO
r_ave = 0.25_dp * r_ave
r2_ave = 0.25_dp * r2_ave
stetra = 1.0_dp - 1.0_dp/(3.0_dp*r_ave*r_ave) * (r2_ave - r_ave * r_ave)
RETURN
END SUBROUTINE compute_tetra_label
SUBROUTINE closest4 (gb0, sorted, npart)
!*************************************************************************************
! subroutine to find the four closest particles of a given species to a given particle
!
! authors: s. chiacchiera, january 2018
!*************************************************************************************
!
! input: - all the coordinates of beads of species "sp" (the others are set to "0")
! - one selected particle of species "sp"
! output: the (ordered by distance) labels of the four closest "sp" particles to it
IMPLICIT none
INTEGER, INTENT(IN) :: gb0, npart
INTEGER, INTENT(OUT) :: sorted (4)
INTEGER :: i, count, ncut, indx, size
REAL(KIND=dp) :: x, y, z, r, volm
REAL(KIND=dp) :: rcut, rmin, sorted_r (4)
REAL(KIND=dp), ALLOCATABLE :: list (:,:)
ncut = 15 !10 ! a bit more than 4, to be safe.
volm = dimx*dimy*dimz
size = MIN (npart - 1, 2 * ncut)
rcut = (0.75_dp/pi * ncut / npart * volm) ** (1.0_dp/3.0_dp)
count = 0
ALLOCATE (list (size, 2))
list = 0.0_dp
DO i = 1, nsyst
IF (i == gb0) CYCLE
IF (ltp (i) /= sp) CYCLE
x = xxx (i) - xxx (gb0)
y = yyy (i) - yyy (gb0)
z = zzz (i) - zzz (gb0)
!!!
! CALL images (xab, yab, zab, dimx, dimy, dimz, srfx, srfy, srfz, shrdx, shrdy, shrdz)
x = x - dimx * ANINT (x/dimx)
y = y - dimy * ANINT (y/dimy)
z = z - dimz * ANINT (z/dimz)
!!!
r = SQRT(x * x + y * y + z * z)
IF (r > rcut) CYCLE
count = count + 1
IF (count>size) THEN
WRITE(*,*) "error: too many particles!"
STOP
END IF
list (count, 1) = REAL(i, KIND=dp) ! store the global index
list (count, 2) = r ! store the distance to gb0
END DO
! WRITE (*,*) "rcut=", rcut ! uncomment to see radius of search region
! WRITE (*,*) gb0, count ! uncomment to see the number of particles within it
IF (count < 4) THEN
WRITE (*,*) "error: fewer than 4 neighbours - ",count," - found! Increase the searched volume (-> ncut)"
WRITE (*,*) "time=",time
STOP
END IF
! sorting by distance
sorted (:) = 0
sorted_r (:) = 0.0_dp
DO j = 1, 4
rmin = rcut
indx = 0
DO i = 1, count
IF ((NINT(list (i,1)) == sorted (1)) .OR. (NINT(list (i,1)) == sorted (2)) .OR. &
(NINT(list (i,1)) == sorted (3)) .OR. (NINT(list (i,1)) == sorted (4))) CYCLE
IF (list (i,2) < rmin) THEN
rmin = list(i,2)
indx = NINT(list(i,1))
END IF
END DO
sorted (j) = indx
sorted_r (j) = rmin
END DO
! WRITE (*,'(4(1x,I6))') sorted ! uncomment to see the labels of nn of gb0 (sorted by distance)
! WRITE (*,'(4(1x,f13.6))') sorted_r ! uncomment to see the corresponding distances
DEALLOCATE (list) ! for fixed size, could allocate/deallocate in the main
RETURN
END SUBROUTINE closest4
END PROGRAM tetrahedral
|
| [Duboue2015] | E. Duboué-Dijon, A. Laage, Characterization of the local structure in liquid water by various order parameters, J. Phys. Chem. B, 119, 8406 (2015). |
| [1] | The angle
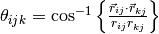 where
where 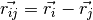 and and 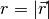 . . |
| [2] | Compilation has been tested with the GNU compiler GCC, version 10.2.0. |
| [3] | The diamond cubic crystal lattice (https://en.wikipedia.org/wiki/Diamond_cubic) is a repeating pattern of
8 atoms. Their coordinates may be given as:
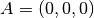 , , 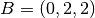 , , 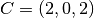 , , 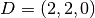 , , 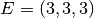 , ,
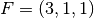 , , 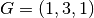 , and , and 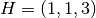 in a unit cubic cell of side in a unit cubic cell of side 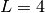 .
One can check that, with the minimum image convention, each
particle has its 4 closest neighbours at a distance .
One can check that, with the minimum image convention, each
particle has its 4 closest neighbours at a distance 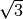 , and all the angles are , and all the angles are
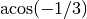 .
For this configuration (also if repeated periodically along the three Cartesian axis), .
For this configuration (also if repeated periodically along the three Cartesian axis),
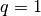 and and 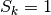 . . |