Caesar electronic structure interface¶
Purpose of Module¶
Calculating vibrational properties requires a mapping of the nuclear potential energy
surface (PES) 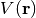 , where
, where  is the collective
coordinate describing the locations of the nuclei. Caesar maps the PES
by sampling it at a number of nuclear configurations
is the collective
coordinate describing the locations of the nuclei. Caesar maps the PES
by sampling it at a number of nuclear configurations 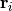 .
.
In software terms, each PES sample represents a single electronic structure calculation,
where the electronic structure code is given the nuclear configuration
, and calculates the value of the PES at that configuration,
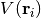 , optionally along with other quantities such as the forces
, optionally along with other quantities such as the forces
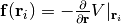 and the Hessian matrix
and the Hessian matrix
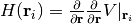 .
.
The Caesar electronic structure interface enables Caesar to be used with a wide range of electronic structure codes, by treating each electronic structure calculation as a black box.
The Electronic Structure Run Script¶
Caesar generates a nested directory structure, with the input file for
each configuration written to its own calculation directory. A
user-provided run script is then called repeatedly, once for each calculation
directory. This script is expected to read any required parameters from the root
directory, read the input file from the calculation directory, and then call the
electronic structure code and write the electronic structure results to an output
file in the calculation directory.
The calculation input and output files can be in a number of formats used by existing electronic structure codes, or they can be in a simple plain-text format.
The plain-text input file, structure.dat is formatted as
Lattice
L_xx L_xy L_xz
L_yx L_yy L_yz
L_zx L_zy L_zz
Reciprocal Lattice
L'_xx L'_xy L'_xz
L'_yx L'_yy L'_yz
L'_zx L'_zy L'_zz
Atoms
z_1 r_1x r_1y r_1z
z_2 r_2x r_2y r_2z
...
z_n r_nx r_ny r_nz
Supercell
S_xx S_xy S_xz
S_yx S_yy S_yz
S_zx S_zy S_zz
Reciprocal Supercell
S'_xx S'_xy S'_xz
S'_yx S'_yy S'_yz
S'_zx S'_zy S'_zz
R-vectors
R_1x R_1y R_1z
R_2x R_2y R_2z
...
R_Nx R_Ny R_Nz
G-vectors
G_1x G_1y G_1z
G_2x G_2y G_2z
...
G_Nx G_Ny G_Nz
End
where:
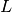 is the supercell lattice matrix, whose rows are the lattice vectors of
the supercell in which the electronic structure calculation should be performed.
is the supercell lattice matrix, whose rows are the lattice vectors of
the supercell in which the electronic structure calculation should be performed.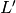 is the reciprocal supercell lattice matrix of the supercell, defined
as
is the reciprocal supercell lattice matrix of the supercell, defined
as 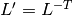 .
.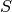 is the supercell matrix, which relates the supercell lattice matrix
to the primitive cell lattice matrix
is the supercell matrix, which relates the supercell lattice matrix
to the primitive cell lattice matrix 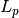 as
as 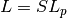 .
.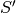 is the reciprocal supercell matrix, defined as
is the reciprocal supercell matrix, defined as 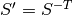 .
.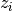 and are the species label and cartesian
coordinate of the
and are the species label and cartesian
coordinate of the 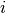 ’th atom.
’th atom.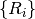 are the R-vectors of the primitive cell which are contained within
the supercell.
are the R-vectors of the primitive cell which are contained within
the supercell.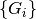 are the G-vectors of the reciprocal supercell which are contained
within the reciprocal primitive cell.
are the G-vectors of the reciprocal supercell which are contained
within the reciprocal primitive cell.- Subscripts
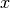 ,
, 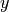 and
and  denote cartesian components.
denote cartesian components.
The plain-text output file, electronic_structure.dat is formatted as
Energy (Hartree):
V
Forces (Hartree/Bohr):
f_1x f_1y f_1z
f_2x f_2y f_2z
...
f_nx f_ny f_nz
Hessian (Hartree/Borh^2):
Atoms: ( 1 1 )
H_11_xx H_11_xy H_11_xz
H_11_yx H_11_yy H_11_yz
H_11_zx H_11_zy H_11_zz
Atoms: ( 2 1 )
H_21_xx H_21_xy H_21_xz
H_21_yx H_21_yy H_21_yz
H_21_zx H_21_zy H_21_zz
...
Atoms: ( n n )
H_nn_xx H_nn_xy H_nn_xz
H_nn_yx H_nn_yy H_nn_yz
H_nn_zx H_nn_zy H_nn_zz
Stress (Hartree/Bohr^3):
sigma_xx sigma_xy sigma_xz
sigma_yx sigma_yy sigma_yz
sigma_zx sigma_zy sigma_zz
where:
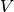 is the energy of the supercell, normalised to energy per supercell.
is the energy of the supercell, normalised to energy per supercell.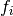 is the force on the ’th atom. The number and order of atom
labels must match those in the input file (i.e. be
is the force on the ’th atom. The number and order of atom
labels must match those in the input file (i.e. be 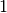 to
to 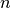 ).
).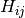 is the block of the Hessian matrix corresponding to atoms
and
is the block of the Hessian matrix corresponding to atoms
and 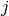 , i.e.
, i.e.
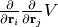 .
.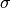 is the stress tensor.
is the stress tensor.- Subscripts , and denote cartesian components.
All sections but Energy are optional. All values must be given in atomic units.
This file is parsed by the ElectronicStructureData class, and the documentation
for this class should be consulted for the full specification of this file. Each line
of the file is split into tokens by whitespace, so the exact whitespace on each line
does not matter as long as there is some whitespace between each token.
Interfaces to Other Electronic Structure Interfaces¶
Rather than interfacing with an electronic structure code directly, Caesar can instead be interfaced with an external package which in turn interfaces with the electronic structure code. Caesar has interfaces to two such packages: QUIP and The Atomic Simulation Environment (ASE).
The QUIP interface needs to be linked at compile
time, as detailed in the
Caesar README.txt file. This is achieved by
setting the CMake configuration option
LINK_TO_QUIP to true.
The ASE interface uses a
python script and does not require additional compilation.
An example script is provided as doc/input_files/example_ase_run_script.py in
the Caesar repository, and this script is
intended to serve as a template for an interface with any of the electronic structure
codes which ASE can interface with.
Source Code¶
The source code for the Caesar electronic structure interface is available from
the src/common/electronic_structure directory of the
Caesar repository