Support of GGA and MGGA functionals from Libxc in FHI-AIMS¶
Libxc provides hundreds of well-tested approaches to calculate interactions between electrons at the atomic scale. Implementing them individually is long and tedious. With this integration, FHI-AIMS has undergone a paradigm shift and greatly expanded its capabilities in this domain.
Purpose of Module¶
FHI-aims is an efficient, accurate all-electron, full-potential electronic structure code package for computational molecular and materials science (non-periodic and periodic systems) using numeric atom-centered basis functions (NAOs) [Blum2009]. The code supports DFT (semilocal and hybrid) and many-body perturbation theory. The numerous implemented exchange-correlation (XC) functionals in the Libxc library extend the possibilities for the users and their simulations in FHI-aims.
The purpose of the module is twofold:
- Implementation of a unified Libxc interface in FHI-aims.
2. Enabling the use of Libxc functionals with the corresponding, properly generated minimal basis functions for GGA and meta-GGA functionals (see detailed information in the Background Information Chapter). This interfaces the scalar- relativistic atomic solver of FHI-aims (default version) with Libxc.
An interface to Libxc has been implemented before, but remained at a “proof-of-concept” stage. Libxc in FHI-aims is not only used for DFT calculations, but is needed for DFPT and the calculation magnetic and optical response properties. After full integration of Libxc, it will form an essential part of each simulation and will be used by most of the FHI-aims user.
In the long term, we hope that Libxc can extend and finally replace the internal FHI-aims XC library helping to move FHI-aims from a monolithic to a more modular software architecture. At the same time, we expect that the Libxc project will benefit from the increase of usability and visibility.
| [Blum2009] | Blum, V., et al. (2009). Ab initio molecular simulations with numeric atom-centered orbitals. CPC, 180 (11), 2175–2196. https://doi.org/10.1016/j.cpc.2009.06.022 |
Background Information¶
While point one from above (Implementation of a unified Libxc interface) is straightforward to implement from the documentation of Libxc the second part needs some further explanation.
The XC potential is needed at two points of a regular DFT calculation in FHI-aims. First, during the initialization generating the minimal basis (i.e. the NAOs) and the initial density. Second, during the usual SCF iterations.
The minimal basis is calculated by solving the scalar-relativistic Schrödinger equation of the free atom for each species. In principle, due to the spherical symmetry of this problem, all contributions of the XC-potentials can be formulated in analytical terms. Thus, this requires a separate routine as during the SCF-cycle. In practice, the one-dimensional radial equations are solved on a dense logarithmic radial grid as described in [Fuchs1999]. An interface to Libxc for this atomic solver and the implementation of the corresponding expressions (the functional derivatives of the XC potential w.r.t the density) were needed. In principle, any XC functional could be used to generate the minimal basis. However, it has been empirically become evident that using the same XC functional for generating the minimal basis and during the SCF iterations guarantees a faster convergence with fewer basis functions – at least for LDA and GGA functionals. This current module only implements the Libxc interface to the various LDA and GGA functionals, but not for the meta-GGAs. Instead, still only the pw-LDA functional is used to generate the basis functions and initial density for all meta-GGA calculations. It is planned to implement a finite-difference approach for generating the meta-GGA minimal basis set in the future as the corresponding analytical expression are becoming more and more numerous (tedious to implement).
| [Fuchs1999] | Fuchs, Martin, and Matthias Scheffler. “Ab initio pseudopotentials for electronic structure calculations of poly-atomic systems using density-functional theory.” CPC 119.1 (1999): 67-98. |
Building and Testing¶
The build of FHI-aims for various compiler and compiler settings is integrated in the FHI-aims Gitlab CI pipeline, where all implemented parts of this module are built, too. A test has been added to the regression test suite of FHI-aims testing the newly implemented interface and the result of a DFT simulation for diamond silicon and the PBE functional.
Source Code¶
Note
The source code of FHI-aims is in principle open, however, a separate license is needed to get access to it. In case you need access, please ask via: mailto:aims-coordinators@fhi-berlin.mpg.de. A brief overview of the needed steps are listed below.
Implementation of a unified Libxc interface¶
This first step was straightforward to implement. Already existing code blocks have been merged into a single module
libxc_interface.f90 and unified to have a consistent interface for all parts of the code. Some effort was needed to
synchronize XC functional dependent runtime variables, which was especially tedious for the range-separated hybrid functional.
All requested resources from Libxc during runtime are denoted in the main output file and citations are given for citation.
Interface to the scalar-relativistic atomic solver¶
Due to the rotational symmetry of the free-atom problem all terms of the XC potential 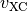 can be express
analytically. The current implementation considers all derivatives up to GGA functionals (here for spin-unpolarized case):
can be express
analytically. The current implementation considers all derivatives up to GGA functionals (here for spin-unpolarized case):
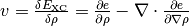
The goal is to express all terms of the energy per volume 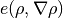 as partial derivatives of the
density or the density gradient. In case of GGA functionals, this is straightforward by using the nabla operator in
spherical coordinates and using the chain rule for the appearing derivatives w.r.t to
as partial derivatives of the
density or the density gradient. In case of GGA functionals, this is straightforward by using the nabla operator in
spherical coordinates and using the chain rule for the appearing derivatives w.r.t to  .
The final expressions have to be transformed in terms of the reduced variables
.
The final expressions have to be transformed in terms of the reduced variables 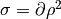 as the
then appearing energy derivatives can be requested from the Libxc library. The implemented routines can handle both
spin-polarized and spin-unpolarized free atoms.
as the
then appearing energy derivatives can be requested from the Libxc library. The implemented routines can handle both
spin-polarized and spin-unpolarized free atoms.