Feedback control mechanism: A Component of the Hierarchical Equilibration Strategy for Polymer Melts¶
The module is an implementation of the existing hierarchical strategy [1] for the equilibration of simple one-component polymer melts in ESPResSO++.
Purpose of Module¶
To study the properties of polymer melts by numerical simulations, equilibrated configurations must be prepared. However, the relaxation time for high molecular weight polymer melts is huge and increases, according to reptation theory, with the third power of the molecular weight. Hence, an effective method for decreasing the equilibration time is required. The hierarchical strategy pioneered in Ref. [1] is a particularly suitable way to do this. The present module provides a part of that method described below.
To decrease the relaxation time, microscopic monomers are coarse-grained (CG)
by mapping each subchain with 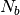 monomers onto a soft blob.
The CG system is then characterized by a much lower molecular weight and
thus is equilibrated quickly. One thus obtains a configuration that is
equilibrated on large scales but does not provide information about
the structure on smaller (i.e. more fine-grained (FG)) scales.
monomers onto a soft blob.
The CG system is then characterized by a much lower molecular weight and
thus is equilibrated quickly. One thus obtains a configuration that is
equilibrated on large scales but does not provide information about
the structure on smaller (i.e. more fine-grained (FG)) scales.
To obtain the latter, the resolution is step-by-step increased by recursively applying a fine-graining procedure to the previous (more coarse-grained) level. In such a fine-graining step, each CG polymer chain is replaced with a more fine-grained chain, by dividing a CG blob into several FG blobs.
In the last step, microscopic monomers are reinserted into CG blobs. This reinsertion procedure is divided into 2 parts. Firstly, monomers are treated as mass points without non-bonded interaction. Starting from this state, repulsive non-bonded interactions are gradually introduced according to the feedback control mechanism explained in Ref. [2]. This procedure makes sure that the final fine-grained conformation is consistent with the conformation at the more coarse-grained level.
The present module provides the python script which performs the feedback control mechanism. The implementation detail is in following below.
The microscopic configuration of
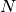 polymers consisted of
polymers consisted of 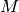 monomers is prepared. The system size
monomers is prepared. The system size 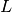 is determined by the number of density
is determined by the number of density 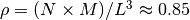 .
. 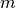 and
and 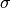 stands for the mass and the diameter of monomers.
stands for the mass and the diameter of monomers.We presupposed that a configuration is already equilibrated at a coarse-grained level and is not equilibrated at a microscopic level.
NVT MD simulation is carried out with bonding potential
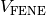 and force-capped-LJ potential
and force-capped-LJ potential 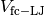 defined as
defined as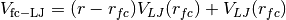 for
for 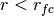 ,
,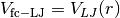 for
for 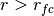 ,
,for preventing too strong repulsive forces. At first we set
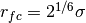 .
.The excluded volume interaction can be gradually introduced with gradually decreasing
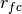 .
In order to obtain the equilibrated structure of polymer melts, is controlled by the difference between the mean-square internal distances of the current configuration and that of the ideal curve in the intermediate region. This difference is defined as
.
In order to obtain the equilibrated structure of polymer melts, is controlled by the difference between the mean-square internal distances of the current configuration and that of the ideal curve in the intermediate region. This difference is defined as![I=\int^{50}_{20} [(<R^2(n)>/n)_{ideal} - (<R^2(n)>/n)_{current}]dn](../../../../../_images/math/b3dcb880d6c4013f370e06743a6978a93c779780.png) ,
,where
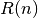 is an internal distance of chain segment of length
is an internal distance of chain segment of length 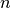 .
For
.
For  , is increased. In contrast, for
, is increased. In contrast, for  , is decreased.
, is decreased.After performing during
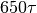 , MD simulation is finished. Where
, MD simulation is finished. Where 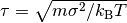 .
.
More detail of this feedback control mechanism is explained in Ref [2].
Background Information¶
The implementation of this module is based on ESPResSO++. You can learn about ESPResSO++ from the following links:
- ESPResSO++ documentation: http://espressopp.github.io/ESPResSo++.pdf
- ESPResSO++ source code: https://github.com/espressopp/espressopp
Building and Testing¶
Explanation of installation:
After installing this module, it can be tested according to the README file found under the following link:
Source Code¶
This module has been merged into ESPResSo++: